|
Inheritance of MTHFR Deficiencies |
|
|
|
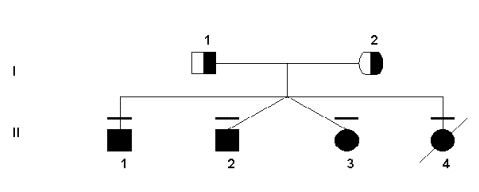
Both the severe and mild deficiencies of MTHFR show autosomal recessive inheritance patterns (Rosenblatt & Erbe, 1977). This work has been supported by pedigree studies, as outlined below. Since the gene for the MTHFR protein was not found using traditional linkage studies, pedigree analysis was not done or used until after its location was already known. However, some research groups constructed pedigrees for families that had MTHFR deficiency in order to link the deficiency to other health problems that may have been transmitted through the generations. For example, the following pedigree was used to verity the transmission of two specific mutations in this particular family.
Sequencing and restriction enzyme digest did indeed show that
the four children were homozygous for both mutations. At the 677bp, most
people are C/C in absence of a mutation. However, the father was T/T
(based on the C to T bp substitution) and the mother was heterozygous
C/T. Although not the only possibility, all of the children born were
homozygous for the mutation, T/T. In addition, the other mutation at the
1081 bp is also C/C wild-type. However, both parents were C/T (again
representing the substitution in the base pairs from C to T).
Unfortunately, the T allele was passed on by both parents to all four of
their children, giving them all the T/T genotype. Interestingly, both
parents had normal health throughout their lives and no noticeable
deficiency in their MTHFR enzyme. Nevertheless, all of their children
have had persistent health problems typical of MTHFR deficiency
(seizures, developmental delays, lack of speech/communication, gait
abnormalities, etc.). Furthermore, their fourth child died at 2 months
of severe pneumonia and respiratory failure, even though the parents had
attempted to do prenatal diagnosis (on chorionic villi and trophoblast),
which only revealed slightly lower MTHFR activity. In analyzing the
different alleles, it was found that the wild-type allele at the 677 bp
generate a 198 bp fragment when digested with Hinf I. However, the C®T substitution creates a new recognition site that splits
that fragment into a 175 and 23 bp fragments (seen in Gel
1). For
the 1081 bp wild-type gene, a Hha I digestion typically yields a 152 bp
fragment. Then, with this C®T
mutation, that Hha I site disappears and the digestion of these alleles
reveals a slightly longer 165 bp fragment (seen in Gel
2).
(Analysis from Tonetti et al 2000). This general algorithm that linked
mutations to altered restriction enzyme recognition sites (and hence,
fragment lengths on gels) was also used in other families to first
determine specific mutations and then to identify them in other
individuals both related and non-related.
|